The sequencing protocol was based on the dideoxy sequencing method (Sanger et al., 1977). In order to sequence from each of the clone inserts in pairs, each plasmid template DNA plate should be equipped with two 384-well cycle sequencing plates. The sequencing reaction used Big Dye Terminator chemistry version 3.1 (Applied Biosystems) and standard M13 or commonly used forward and reverse primers. The sequencing reaction was established by a Biomek FX (Beckman) pipetting station. The robotic arm is responsible for aliquoting the template sample and mixing it with the reaction solution. The reaction solution contains dideoxynucleotides, fluorescently labeled nucleotides, Taq DNA polymerase, sequence primers and buffer.
The template and plate have barcodes and barcode reader tracking on the BiomekFX pipetting station to ensure no errors in template and reaction transfer. The 30-40 linear amplification step was continuously performed in MJ Research Tetrads or 9700 Thermal Cycler (Ap-plied Biosystems). The reaction product can be precipitated with high efficiency at room temperature with isopropanol, stored or suspended in water at 4 C. If the sequencing instrument is normal, a sample film will be automatically generated for each reaction plate after scanning the barcode of the reaction plate. The plate was then transferred to an ABIPrism 3700 DNA Analyzer or an Applied Biosystems 3730xi DNA Analyzer for electrophoresis. The current multimers and software allow for 8 electrophoresis per day on the ABIPrism 3700 DNA Analyzer and 12 times on the Applied Biosystems 3730xl DNA Analyzer with a commissioning time of less than 1 h.
High-throughput sequencing equipment that runs a large number of jobs in parallel typically requires automated management through a laboratory information management and sample tracking system (LIMS) (Kerlavage et al., 1993). At TIGR, this system includes a complete suite of software from early sequencing to end-of-sequence tracking of library construction. After this processing, the data is saved in a Sybase relational database table. The database stores and links all of the data collected throughout the genome sequencing process, allowing the user to backtrack the data stream in a variety of ways, backtracking from the annotated genes to the original sequencing trace files of the genes. This system includes client/server applications for sample management, data entry, library management and sequence processing. After years of improvement, combined with new laboratory methods, new instruments and software, this system has matured and stabilized. These integrated applications include automated vector removal, identification and masking of repetitive elements, discovery of contaminated clones and tracking clones, and template information.
In the sequencing process, the quality of the generated templates and sequences can be systematically monitored daily through a user-friendly interface. This ensures that potential problems in the work are quickly discovered and corrected. Typically, quality control/quality assessments (QC/QA) groups apply quality inspection standards together. They are responsible for inspecting and providing reagents to the production team, and detecting the quality of the template, the failure of the investigation and deviation from the normal performance range in the process, monitoring the quality of the data, auditing, identifying areas for improvement, and making control documents (standard operating procedures) to Ensure that these files are format consistent and technically accurate.
High-throughput sequencing faces five major challengesAs a hot technology in the medical and health industry, gene sequencing has been increasingly recognized in clinical practice in recent years and has been gradually applied to various fields. Especially after the concept of precision medicine was put forward, gene sequencing was favored, and it solved many unknown problems for precision medicine.
Nowadays, gene sequencing has formed a certain industrial scale, and a large number of enterprises have entered different forms. However, behind the rapid development of the surface, there are still a lot of technical challenges. Foreign media "GEN" Dr. Shawn C. Baker wrote an article explaining the difficulties and challenges facing the field. Lei Feng Network (Public No.: Lei Feng) AIHealth column compiled as follows:
Over the past decade, high-throughput sequencing technology has undergone a leap-forward development, with sequencing capabilities rising dramatically and costs falling, both of which are orders of magnitude. So far, there are more than 10,000 sequencing equipment in the world.
Over the past decade or so, major platform companies have focused on improving the ease of use of their systems. Illumina's newest desktop systems, such as the NextSeq, MiSeq, and MiniSeq systems, are operated with kits to reduce the number of manual operations and boot times.
Illumina's systems have been used to make the Ion Torrent system easier to use, but the latter's latest system, the Ion S5, is designed to simplify the entire workflow, from designing the library to data generation.
After listening to many advances in the sequencing industry, such as strong sequencing capabilities, lower cost, and better ease of use, readers outside the industry may mistakenly believe that all the difficulties in gene sequencing have been solved, all of the sequencing process. The obstacles have been removed.
But the real difficulties are just beginning, and a lot of challenges are ahead.
Sample qualityOne of the most problematic areas is also easily overlooked: sample quality, although the test platform is often calibrated and the samples used are calibrated, but real-world samples often face many unexpected challenges.
One of the most commonly used sample types in human gene sequencing is FFPE (formalin-fixed paraffin-embedded). There are many reasons for the widespread use of FFPE, the most important of which is richness. It is estimated that more than 10 billion FFPE samples are archived worldwide. The clinical sample storage of FFPE blocks has become a standard practice at the industrial level, and the number of samples will continue to grow.
In addition to a wide range of applications worldwide, FFPE samples often contain a large amount of phenotypic information available. For example, FFPE samples can be combined with treatment methods and clinical data.
However, the problem with the FFPE sample is that both the fixed process and the storage conditions cause a large amount of DNA damage.
Dr. Hans G. Thormar, CEO and co-founder of BioCule, believes that
After evaluating more than 1000 samples of BioCule's QC platform, we saw a large number of variations and various types of damage in DNA samples, such as interchain, intrachain cross-linking, single-stranded DNA polymerization, and single-stranded DNA disruption.
The number and type of variations in DNA damage, if ignored, may have a negative impact on the final outcome.
Thormar believes that
The impact on downstream applications such as sequencing is enormous: from the failure of simple sequencing library construction to the generation of false libraries, which ultimately leads to errors in results. Therefore, it is critical to properly assess the quality of each sample at the beginning of the sequencing project.
Although major sequencing platform companies have made great efforts to reduce the cost of generating raw sequences, they are not the same in building sequencing libraries. The construction of sequencing libraries for human gene sequencing, which cost approximately $50 per sample, is a relatively small fraction of total spend. But in other applications, such as bacterial genome sequencing or low-depth RNA sequencing, it accounts for a large portion of the total cost.
Several groups have studied diversified home-made solutions and expect to be able to effectively reduce costs, but there has not been much development in the commercial sector. One of the highlights in the development of single-cell sequencing solutions, such as the 10X Genomics ChromiumTM system, allows hundreds to tens of thousands of samples to be processed in parallel using a bead-based system.
Dr. Serge Saxonov, CEO and co-founder of 10X Genomics, insists that
We believe that single-cell RNA sequencing is the right way to perform gene expression analysis. In the next few years, in many parts of the world, RNA testing will shift to single-cell resolution, and our platform is likely to lead the wave in this area.
For large projects, such as reducing sample costs, the highly versatile solution required for single-cell RNA sequencing will be a key factor.
Long readings and short readingsIllumina's dominance in the gene sequencing market means that most of the data generated so far is based on short reads (short reads), the sequence produced by the high-throughput sequencing platform is called reads, which is the sequence of sequence reads. , the smallest unit of sequencing). The production of a large number of short readings is suitable for most applications. For example, detecting single nucleotide polymorphisms of genomic DNA and counting RNA transcripts. However, in many other applications, only short readings are not sufficient, such as reading highly repetitive regions of the genome and determining long chain structures.
Long reading platforms, such as RSII and Sequel from Pacific Biosciences, and MinION from Oxford Nanopore, typically produce readings in the 15-20 kb range, up to 100 kb in length. Such a platform has won the appreciation of the scientific community, such as Dr. Charles Gasser, professor of cell biology at the University of California, Davis.
I was impressed with the success of genome assembly with long reading methods, especially in hybrid assemblies when combined with short reading high fidelity data. The combination of technologies enables small groups, small budget individual researchers to produce a usable assembly from a new biological genome.
In order to take full advantage of these long reading platforms, it is necessary to prepare DNA samples by new methods. Standard molecular biology methods have not been optimized for the separation of ultralong-length DNA fragments, so care must be taken when preparing long reading libraries.
For example, suppliers have created a high-molecular-weight kit for isolating DNA fragments larger than 100 kb, optimizing a targeted DNA approach to selectively enrich large fragments of DNA, and in order to maximize the yield of long reads, these methods and techniques Must be mastered.
A special form of short reading is a link reading, such as 10X Genomics, which can be used as an alternative to true long readings. The link readings are generated as follows: each long DNA segment, usually larger than 100 kb, contains a unique barcode for each short reading produced. This unique barcode links the separated short readings during the analysis phase. Together, it provides long-chain genetic information, making it possible to construct large haplotype blocks and to interpret complex structural information.
Short reading sequencing, due to its high precision and high throughput, usually has powerful functions, but only a small amount of genetic information can be obtained. This is because the genome is essentially repetitive and a large amount of information in the genome is encoded in the long chain.
data analysisOne of the big challenges for researchers is that the amount of data generated is very large. A single 30X human genome-wide sample has a BAM file (semi-compression alignment file) of approximately 90 GB; a relatively medium-sized project containing 100 samples with a BAM file size of 9 TB.
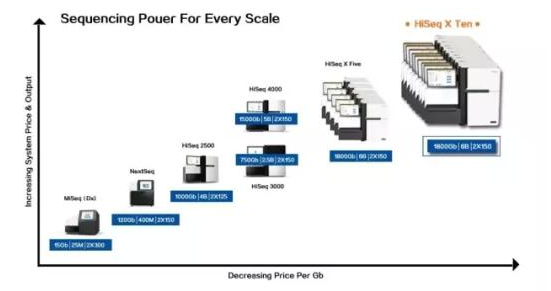
An Illumina HiSeq X instrument can generate more than 130TB of data per year, and quickly storing data becomes a big issue. For example, the Broad study analyzed a 30X human genome-wide rate-generating gene sequencing data every 12 minutes—approximately 4000 TB of BAM files per year.
BAM files can be converted to VCF files (variant call format), which only contains information that is different from the standard sequence. Although the VCF file is small and more usable, it is still necessary to save the original sequence file, which is convenient for researchers to view the data in the future.
As the cost of sequencing declines, some people have come to the conclusion that resequencing samples can be easy and potentially cheaper, and researchers have a lot of room to choose when analyzing large amounts of data. But in fact, there are more than 3,000 sequence analysis tools available in OMICtools, and it's not easy for researchers to find the best one.
Finally, there is a challenge for clinical samples: providing consistent and reliable interpretation of the variation of sequencing sequences.
A typical exon contains 10,000 to 20,000 mutations, and a whole gene sample produces more than 3 million mutations. In the usual interpretation, the disease similarity classification is based on the variation.
To assist in guiding clinicians, the American Society of Medical Genetics and Genomics, the Molecular Pathology Association, and the American College of Pathologists have created a system for classifying mutations. The catalogue includes pathogenicity, possible pathogenicity, and uncertain saliency (currently accounting for the vast majority of exogenous and whole-genome samples), which may be benign and benign.
However, this option has its limitations. Instantly use the same classification scheme to classify the same database, and different project groups may present different interpretations. In a pilot study of the new system, the interpretation of the classification was consistent in only 34% of the different clinical laboratories involved.
If there is a disagreement or if additional analysis is needed to interpret the results of the experiment, there is a problem with reimbursement. Reimbursement for NGS-based testing can be a major obstacle, but reimbursement for explanations is almost impossible.
Dr. Jennifer Friedman, clinical researcher at the Rady Children's Genome Medicine Institute, said
It is impossible for the laboratory to pay for the interpretation of the trial. If this service is available, it is very valuable, but no one does it.
There is no way to pay for this, insurance companies are not reimbursed. Despite the increased attention to precision medicine, no explanations made by clinicians or laboratories have been recognized or valued by health care payers.
So far, the analysis of patient samples has been largely treated as a research project, an option in research hospitals and used only for a limited number of patients.
Desktop Multi-function Adapter is suitable for any normal brands Laptop. Wall Multi-function Adapter is suitable for people who always travel any country. Its plug is US/UK/AU/EU etc. We can produce the item according to your specific requirement. The material of this product is PC+ABS. All condition of our product is 100% brand new.
Our products built with input/output overvoltage protection, input/output overcurrent protection, over temperature protection, over power protection and short circuit protection. You can send more details of this product, so that we can offer best service to you!
Multi-Function Adapter, 12W Wall Adapter, 30W Wall Adapter ,90W Desktop Adapter
Shenzhen Waweis Technology Co., Ltd. , https://www.huaweishiadapter.com